MegaSSR is a robust online server that identifies Simple Sequence Repeats (SSR) and enables the design of SSR markers in high-throughput data. MegaSSR perfectly matches any target genome (including Plantae, Protozoa, Animalia, Chromista, Fungi, Archaea, and Bacteria). The pipeline is freely available at https://bioinformatics.um6p.ma/MegaSSR and github repository.
The computational pipeline consists of the SSR identification tool MISA, Primer3 for PCR primer design, Primersearch (EMBOSS) for in silico validation of designed SSR primers, and Bandwagon for in silico gel visualization. Other processes, such as SSR classification, annotation, and motif comparisons, were performed using custom scripts written in various programming languages such as Perl, Python, and shell script.
MegaSSR running with two options:
1: Simple Sequence Repeat identification, classification, SSR marker design, in silico validation of the designed SSR primers, and gel visualization.
2: Simple Sequence Repeat identification, classification, gene-based annotation, motif comparison, SSR marker design, in silico validation of the designed SSR primers, and gel visualization.
MegaSSR has been tested on Ubuntu 18.04 and 20.04.
- Install
- Required data
- Usage
- Run Example
- Output files
- Output files example
The installation require conda. You can install all dependencies for running MegaSSR in a new conda environment using the MegaSSR.yml and plots.yml files. If you do not have conda, please follow this tutorial.
1- Download repository from github
git clone https://github.com/MoradMMokhtar/MegaSSR.git
2- Go to the MegaSSR folder
unzip MegaSSR-main.zip
cd MegaSSR
3- Create the MegaSSR environment with dependencies
conda env create -f MegaSSR.yml
| Data | Option 1 | Option 2 |
|---|---|---|
| Genome sequence - Fasta file with chromosomes/scaffolds/contigs sequences | Required | Required |
| Genome annotation - GFF/GFF3 file with genome annotations (gene,CDS,mRNA) | Not Required | Required |
Go to the MegaSSR folder
bash MegaSSR.sh -A [1 or 2] -F [Genome in FASTA Format] -G [GFF/GFF3 File]
#Required arguments:
-A The analysis type [1 or 2]
1 (for Simple Sequence Repeat identification, classification, and SSR marker design 'This analysis needs FASTA file only')
2 (for Simple Sequence Repeat identification, classification, gene-based annotation, motif comparison, and SSR marker design 'This analysis needs FASTA and GFF files')
-F Your path to the genome sequence (Fasta file).
-G Your path to the genome annotation (GFF/GFF3 file). Required with argument -A 2 only.
#Optional arguments:
-P Outfileprefix, default is results.
-1 Mininum number of Mononucleotide motifs, default is 10
-2 Mininum number of Dinucleotide motifs, default is 6
-3 Mininum number of Trinucleotide motifs, default is 5
-4 Mininum number of Tetranucleotide motifs, default is 4
-5 Mininum number of Pentanucleotide motifs, default is 3
-6 Mininum number of Hexanucleotide motifs, default is 3
-C Maximum difference between the two motifs by base pairs, default is 100
-s Mininum primer length, default is 18
-S Maxinum primer length, default is 22
-O Optimal primer length, default is 18, default is 20
-R PCR product size range, default is 100-500]
-v Print MegaSSR version and exit
-B Calculate the number of alleles for each SSR primer and plot the migration patterns of the DNA bands [yes|no], default is no"
-L The maximum length by base pair for allele search, default value is 1000"
-I The maximum number of primers in each image. Note that only primers with more than one band are drawn. The default value is 50"
-h Print this Help
Default parameters:bash MegaSSR.sh -A 2 -F /path/to/genome_fasta_file -G /path/to/gff_file -P results -1 20 -2 10 -3 9 -4 5 -5 4 -6 4 -C 100 -s 18 -O 20 -S 22 -R 100-500 -B no -t 4 -L 1000 -I 50
bash MegaSSR_For_Test.sh -A 2 -G NC_003070.9_Arabidopsis_thaliana.gff.zip -F NC_003070.9_Arabidopsis_thaliana.fna.zip -P test -1 20 -2 10 -3 9 -4 5 -5 4 -6 4 -C 100 -s 18 -O 20 -S 22 -R 100-500 -B no -t 4 -L 1000 -I 50
We have collected the main output files in the Results folder in the main output directory. The results are presented in the form of tables and images as follows:
| # | File name | Option 1 | Option 2 |
|---|---|---|---|
| 1 | Identified SSR motifs table.csv | ✓ | ✓ |
| 2 | The distribution of the different SSR classes.csv | ✓ | ✓ |
| 3 | Summary of identified SSR motifs.csv | ✓ | ✓ |
| 4 | Frequency of the identified SSR motifs.csv | ✓ | ✓ |
| 5 | Frequency of identified SSR motifs considering complementarity.csv | ✓ | ✓ |
| 6 | Desinged genic SSR primer.csv | X | ✓ |
| 7 | Genic SSR primer statistics.csv | X | ✓ |
| 8 | SSR primer statistics.csv | ✓ | X |
| 9 | Desinged SSR primer.csv | ✓ | X |
| 10 | Genic SSR repeats with annotations.csv | X | ✓ |
| 11 | non-genic SSR repeats with annotations.csv | X | ✓ |
| 12 | Desinged non-genic SSR primers.csv | X | ✓ |
| 13 | Statistics of non-genic SSR primers.csv | X | ✓ |
| 14 | Motif sequences shared between genic and non-genic regions.csv | X | ✓ |
| 15 | Reprate sequences Unique to genic regions.csv | X | ✓ |
| 16 | Reprate sequences Unique to non-genic regions.csv | X | ✓ |
| 17 | Common reprate between genic and non-genic regions.png | X | ✓ |
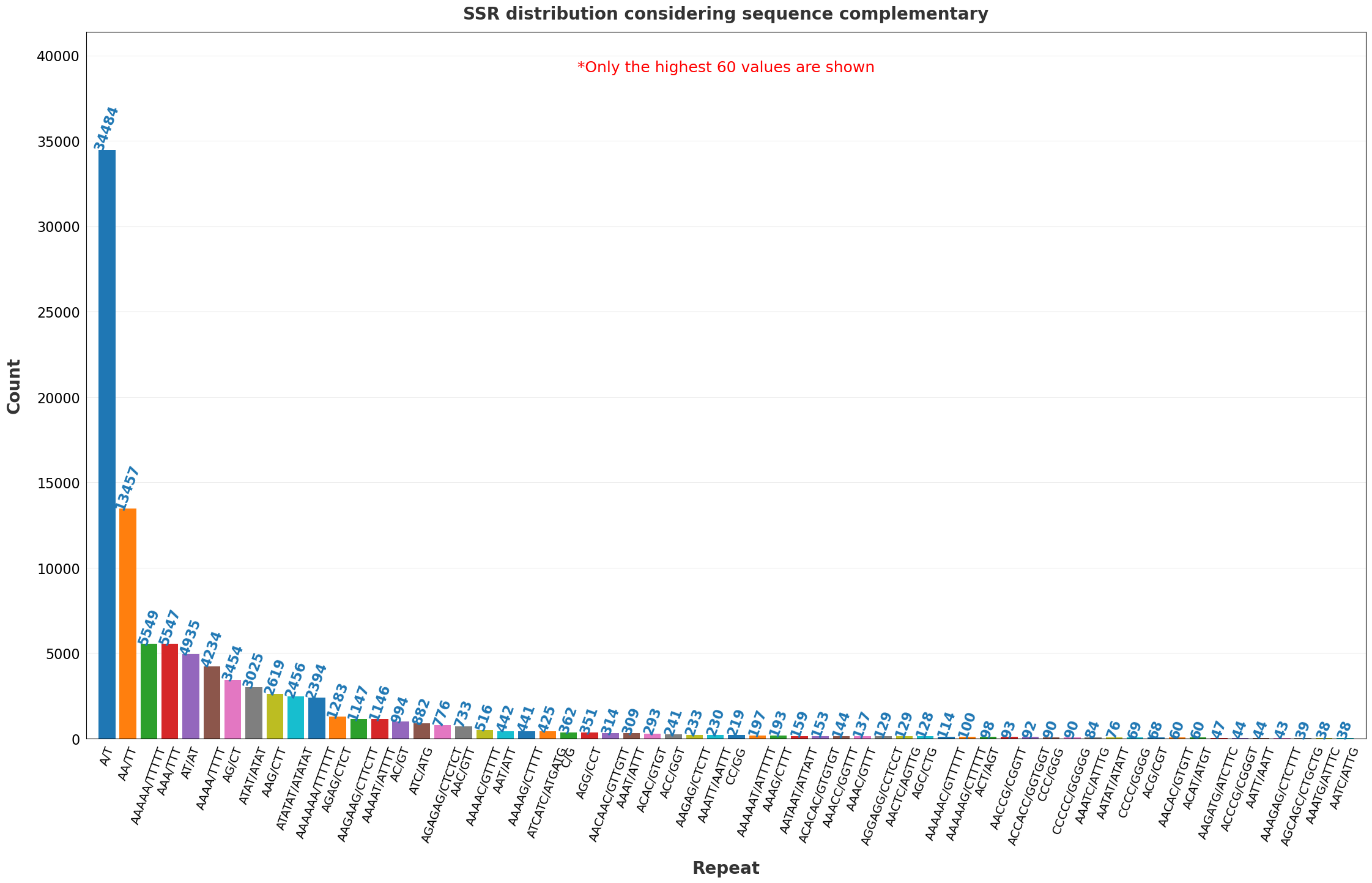
| 18 | SSR distribution considering sequence complementarity.png | ✓ | ✓ |
| 19 | Unique repeats in the genic region.png | X | ✓ |
| 20 | Unique repeats of the non-genic region.png | X | ✓ |
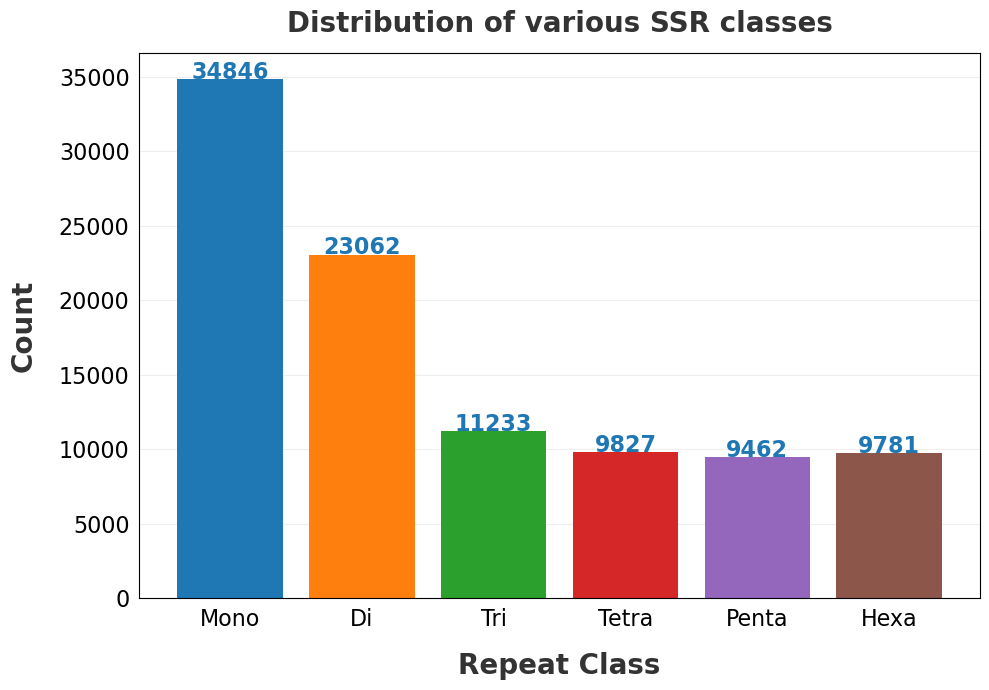
| 21 | Distribution of the different SSR classes.png | ✓ | ✓ |
| 22 | Frequency of the identified SSR motifs.png | ✓ | ✓ |
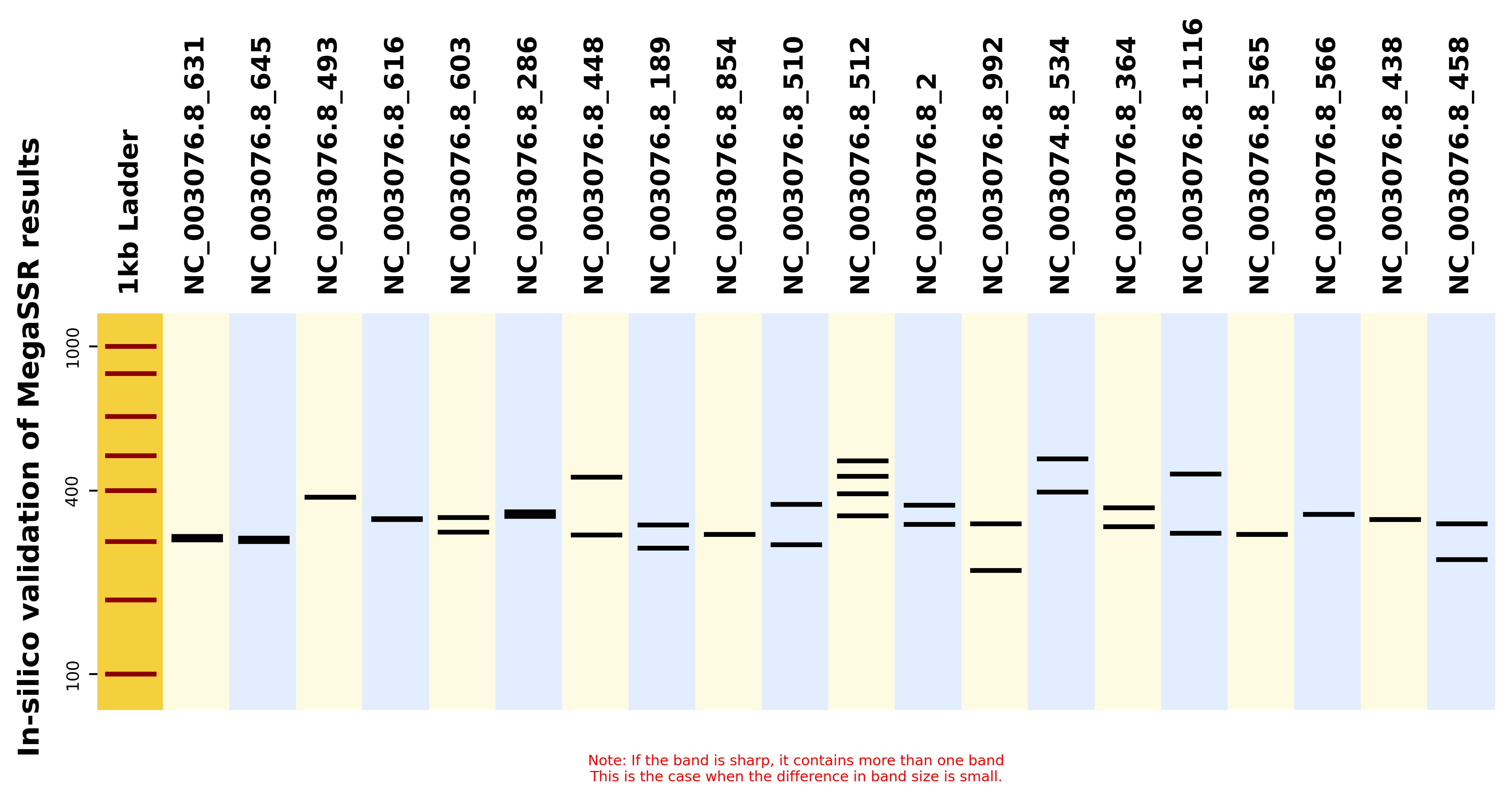
| 23 | x_insilco_gel.jpg | ✓ | ✓ |
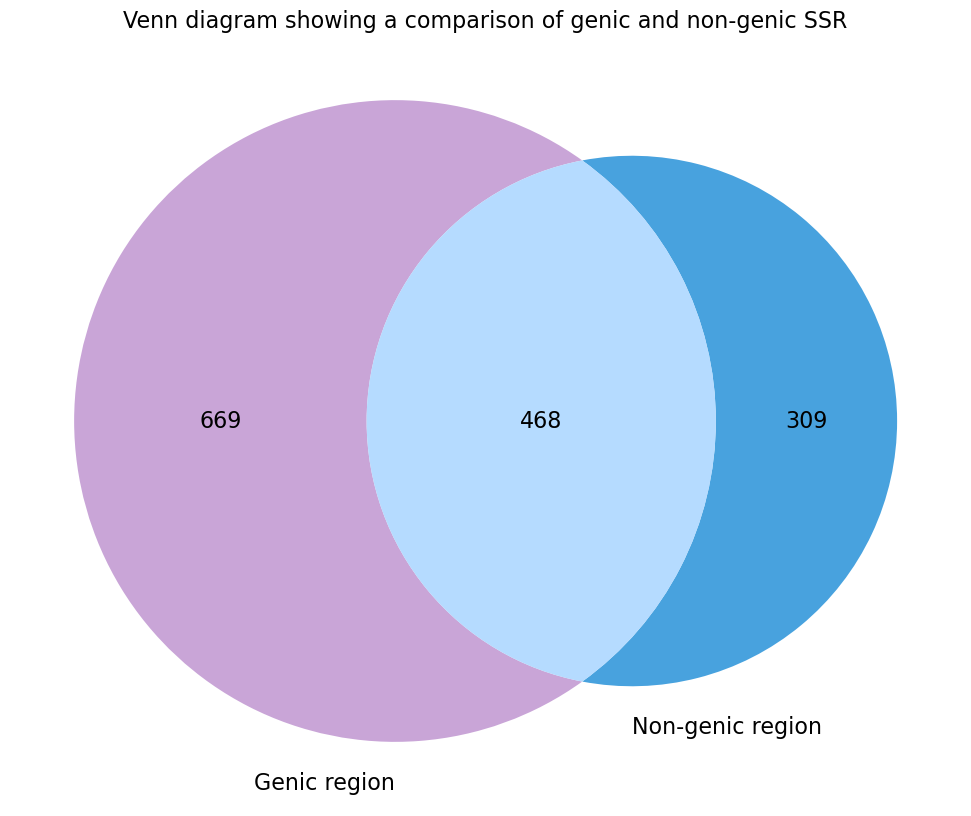
| 24 | Venn diagram of genic and non-genic SSR.png | X | ✓ |
| 25 | SSR primer alleles.csv | ✓ | ✓ |
| 26 | Genic SSR flanking regions.fa | X | ✓ |
| 27 | Non-genic SSR flanking regions.fa | X | ✓ |
| 27 | SSR flanking regions.fa | ✓ | X |
-
In silico gel visualization
-
Venn diagram of identified genic and non-genic SSR
-
Distribution of the different SSR classes
-
SSR distribution considering sequence complementarity
-
Frequency of the identified SSR motifs
-
Common reprate between genic and non-genic regions
-
Unique repeats in the genic region
-
Unique repeats of the non-genic region
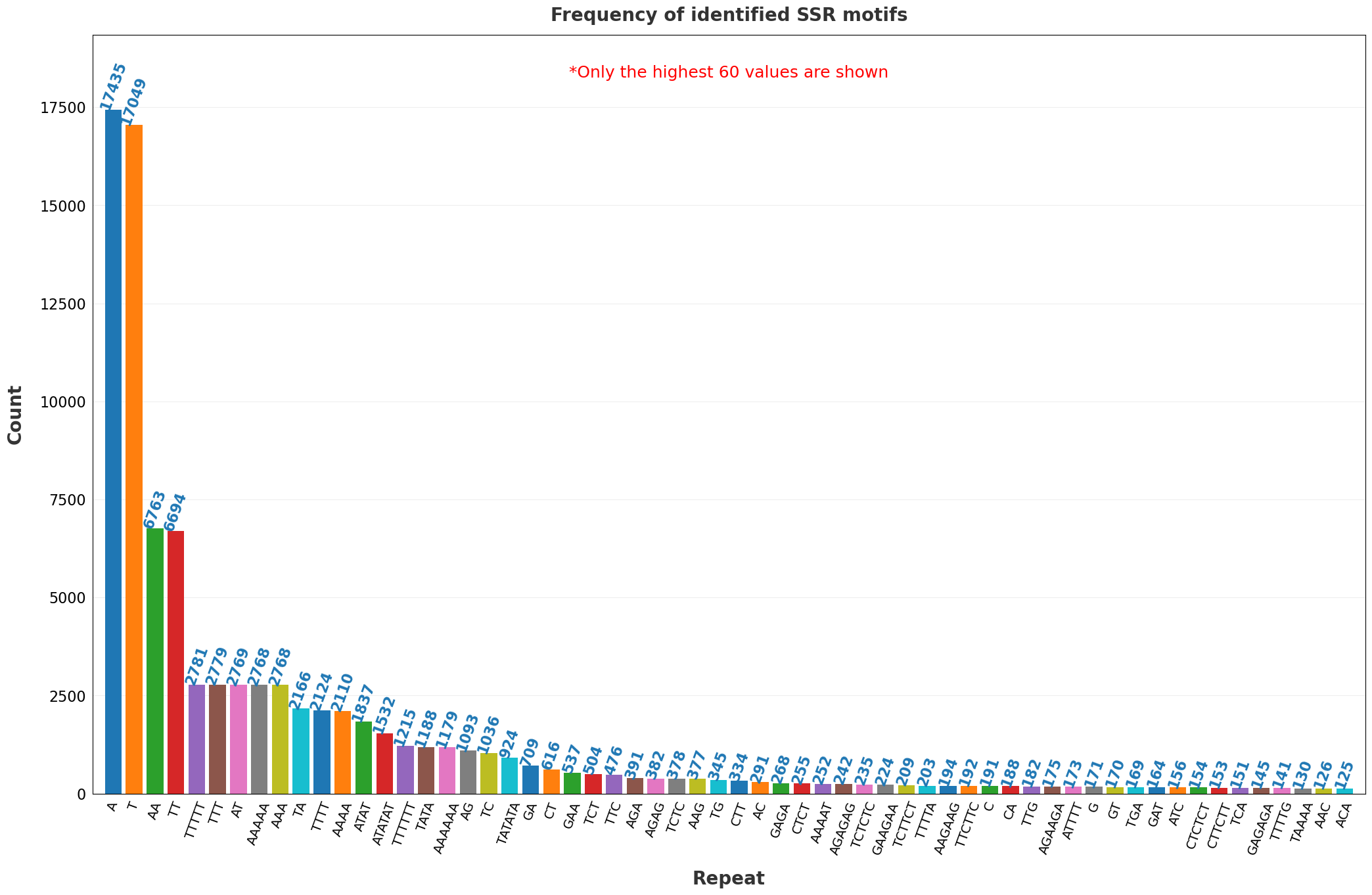
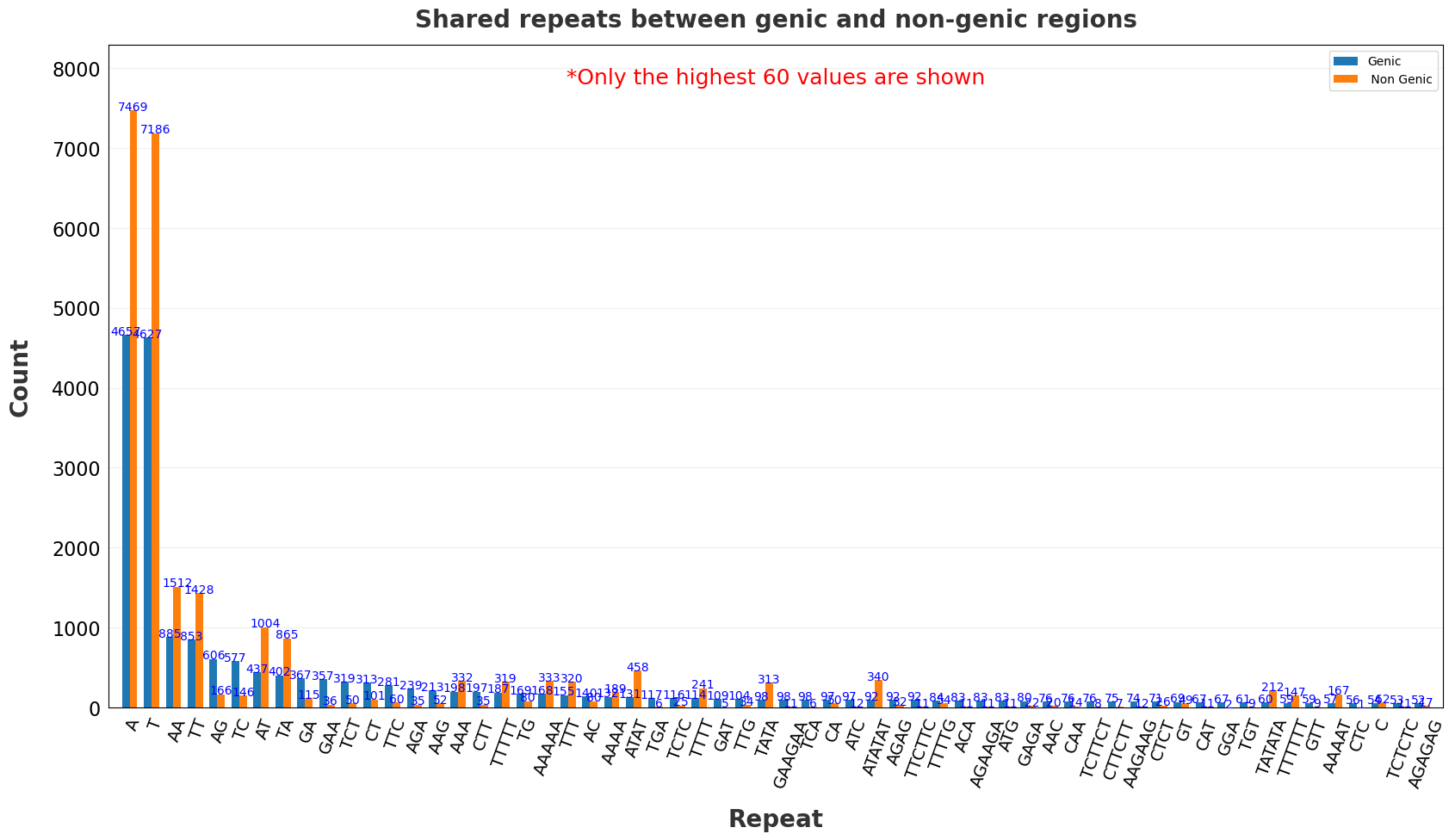
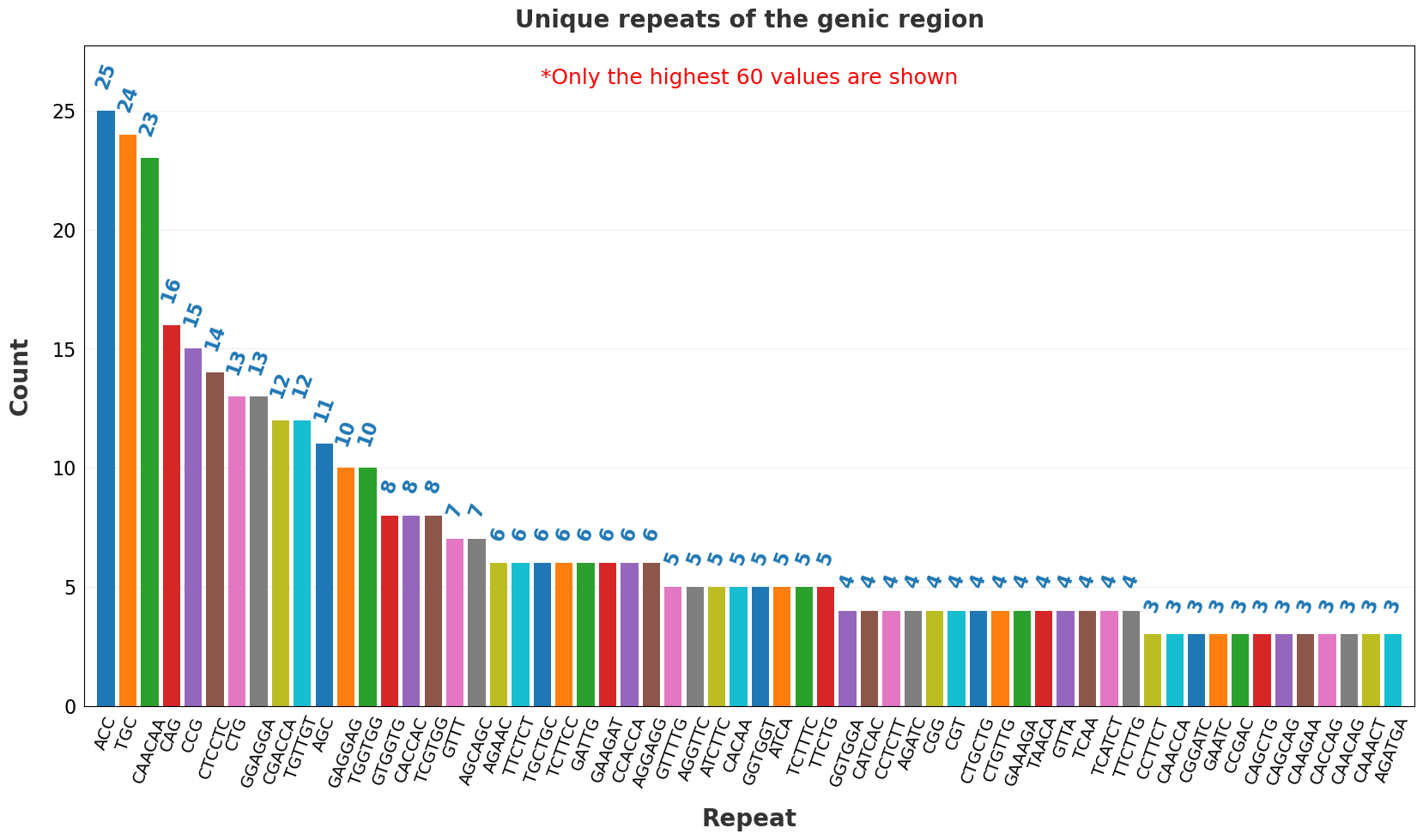
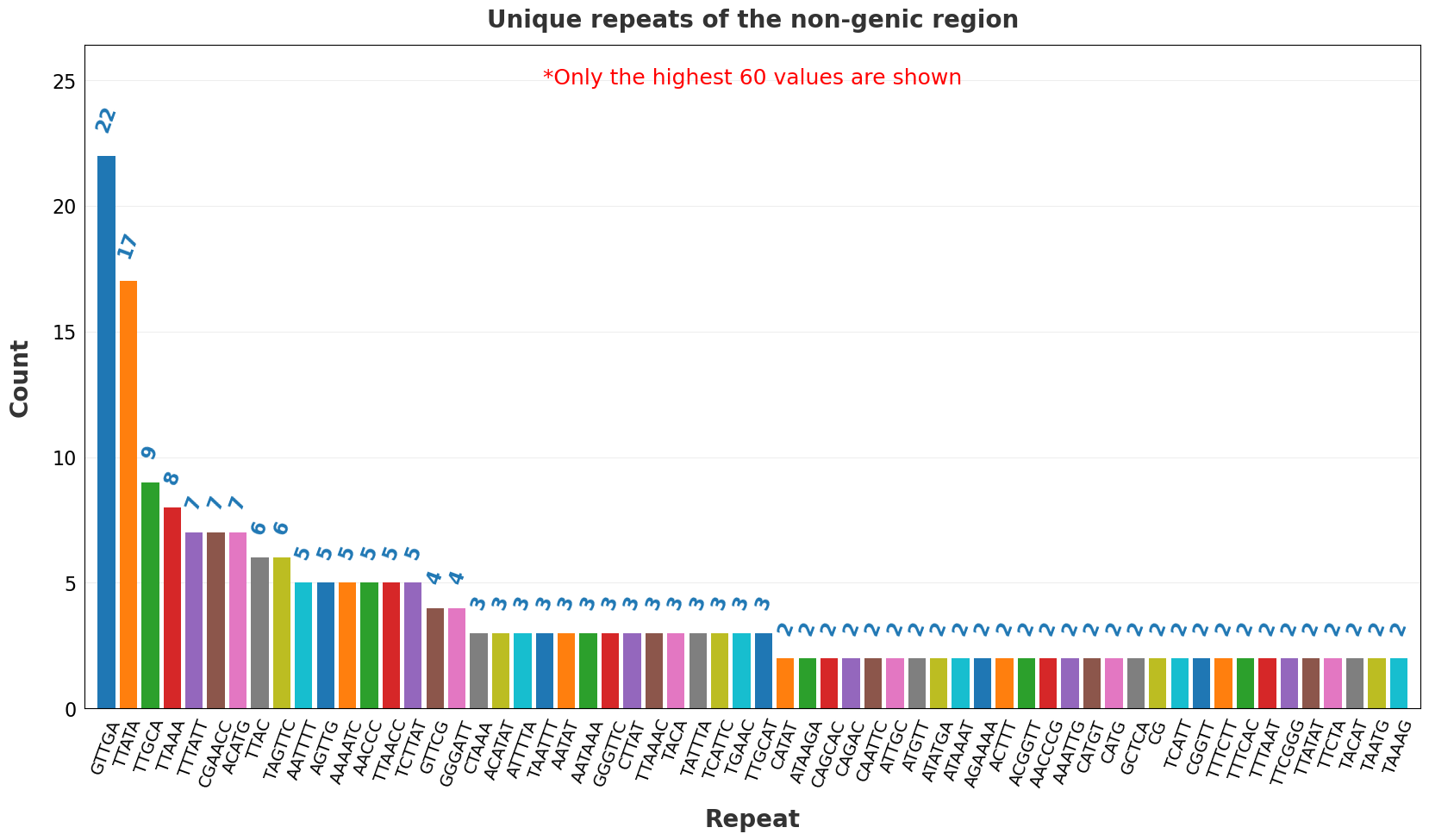
To report bugs and give us suggestions, you can open an issue on the github repository. Feel free also to contact us by e-mail (morad.mokhtar@ageri.sci.eg); (achraf.elallali@um6p.ma).
Morad M. Mokhtar, Alsamman M. Alsamman and Achraf El Allali (2023). MegaSSR: A webserver for large scale SSR identification, classification, and marker development. Frontiers in Plant Science, 14, [https://doi.org/10.3389/fpls.2023.1219055]